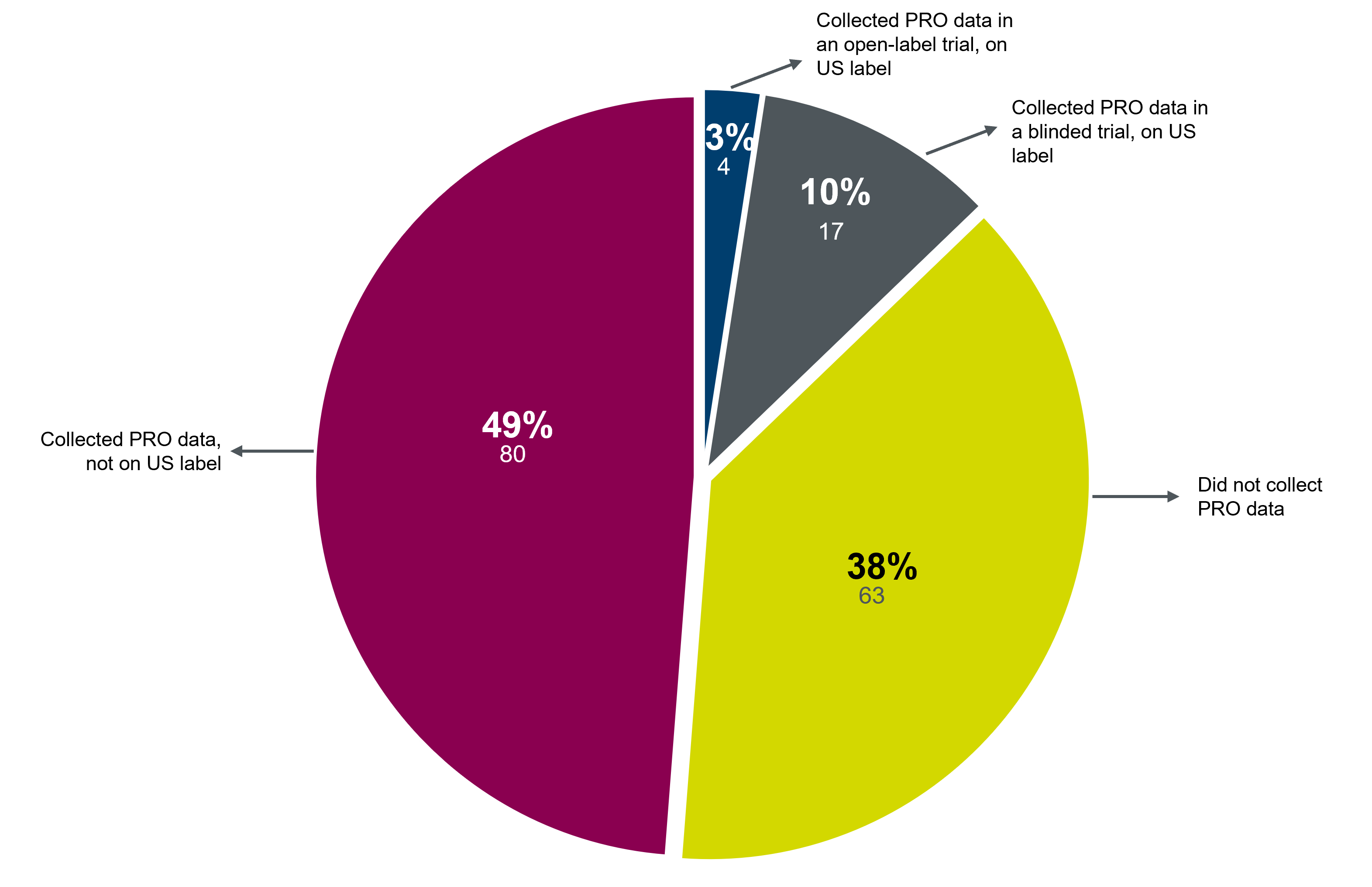
PRO instruments must have “content validity,” meaning they must be relevant and specific to the disease being studied. That requires a clear understanding of the natural history of the disease, the key symptoms that may improve from treatment, and an appropriate length of time to see a clinical benefit. Specify and define concepts—such as signs, symptoms, and impacts—important to the target patient population and likely to demonstrate meaningful and interpretable changes in clinical trials. Seek input from patients and expert clinicians.
It takes time—sometimes years—and effort to do this right. At Parexel, we are currently working with a sponsor to design a PRO for a rare pediatric central nervous system disorder. Clinical trials of this disease traditionally use the primary endpoint of reducing seizures. But seizure control is not the only symptom critical to quality of life in this setting: these children often cannot feed themselves or verbalize, functions that families, caregivers, and patients prize. So, we are developing a new PRO to quantify changes in self-feeding and verbalization. We have worked closely with patients and caregivers and scoured the medical literature to determine what would represent a meaningful change in vocalization and how to measure it precisely. We are combining modules from multiple already-validated PROs into a novel instrument we can validate in this disease and use in clinical trials.
Initially, we will use the new PRO as a secondary or exploratory endpoint in a Phase 1/2 trial. If successful, we will use it for the pivotal trial. We hope to gain FDA guidance and acceptance of this approach during Type B and C meetings. Developing a new PRO requires careful planning, strong rationale, and high-quality evidence.
Sponsors must also ensure that the patient population can validly and reliably self-report (some patients may be too young or sick) and select a PRO that accommodates a spectrum of cognition and mobility levels. Also, make sure the recall period for a PRO is at most two weeks. Otherwise, the FDA may question the validity of the results.
For statistical validity and comparability, the mechanism used to report PRO data should be consistent throughout a trial. A drug recently approved by the FDA for an orphan metabolic disorder used a daily questionnaire to measure patients’ hunger levels. However, partway through the pivotal study, patients switched from answering the questions on paper to using an e-diary. The sponsor did not subject the new e-diary device to usability testing or conduct patient cognitive interviews to assess its functionality, questionnaire comprehension, and ease of use. The FDA concluded there was insufficient data to demonstrate that patients understood one of the questions. Although the PRO data, in this case, made it onto the label (the PRO was the primary endpoint of the study), it’s a cautionary tale for other sponsors.