Lung cancer, primarily non-small cell lung cancer (NSCLC), remains the most common and deadly cancer worldwide.1 Since 2020, the FDA has approved 43 new drugs to treat lung cancer. Yet, patients with advanced disease still face significant unmet needs.2 Major pharmaceutical companies and emerging biotechs are racing to develop the next generation of NSCLC drugs.
However, two recent advances in basic science and regulatory policy impact sponsors. First, scientists have long recognized NSCLC as a group of related but distinct diseases rather than a single histological entity. This understanding has expanded as the remarkable molecular and cellular diversity of NSCLC, highlighted by numerous biomarkers, has become clearer. Developers must therefore decide early in the development process whether to target specific biomarker-driven subgroups or all patients. Second, the FDA’s Project Optimus, launched in 2021, requires comprehensive dose optimization for every anticancer drug.
In the highly competitive NSCLC therapeutic landscape, adaptive trial designs and adaptive decision-making are increasingly essential for accelerating development and ensuring commercial viability.
Slow down to speed up
The FDA expects that thorough dose optimization will benefit patients by ensuring that cancer drugs are more effective and safer. Recent studies have found that treating patients with lower doses of anticancer drugs may reduce toxicities, allowing patients to remain on treatment longer and ultimately leading to improved efficacy.3
Before Project Optimus, the goal of dose escalation was to find the maximum tolerated dose (MTD). The goal now is to determine a range of effective doses for further optimization. Dose optimization requires sponsors to enroll more patients, gather more data, conduct additional analyses, and allocate more time and resources to early-stage trials. The new requirements for dose optimization can be challenging for small biotechs with limited funding and in-house expertise.
At Parexel, we advise sponsors to use an integrated, adaptive first-in-human trial design that mitigates risks by systematically removing the uncertainties at each stage of early drug development: target validation, disease selection, and dose optimization.
Recently, we met with a biotech company looking for a simple dose-escalation trial to show progress and attract funding. They thought that an adaptive Phase 1-2 trial with escalation, optimization, and expansion was too complicated and slow. However, this was shortsighted because the FDA later required a more complex adaptive design with multiple arms and doses. Starting with a seamless, adaptive trial from the beginning saves time overall. Failing to comply with the FDA’s Project Optimus could lead to years-long delays in drug development. Spending a few months designing a data-rich trial reduces risks and speeds up development, which can significantly increase an asset’s value. Informed investors understand the importance of a comprehensive drug development plan that includes dose optimization.
Key elements of adaptive trial designs
Efficient study designs can reduce the time spent on dose escalation, optimization, and expansion by combining proof-of-concept and dose-finding. Successful adaptive trials in early-phase NSCLC incorporate six key elements:
- Initial broad patient cohorts: Early trials may start with a broad "all comers" approach for solid tumors before narrowing the focus to NSCLC and then to specific biomarker-defined subgroups as data accumulates. This enables researchers to assess safety and tolerability, and explore the drug's effect across various populations, potentially avoiding missteps in defining patient populations and biomarker strategies too early or narrowly. Recently, we worked on such a Phase 1-2 trial that began by enrolling patients with solid tumors and progressively narrowed its focus to NSCLC based on accumulating data. The patient population was ultimately refined to include only those with specific mutations.
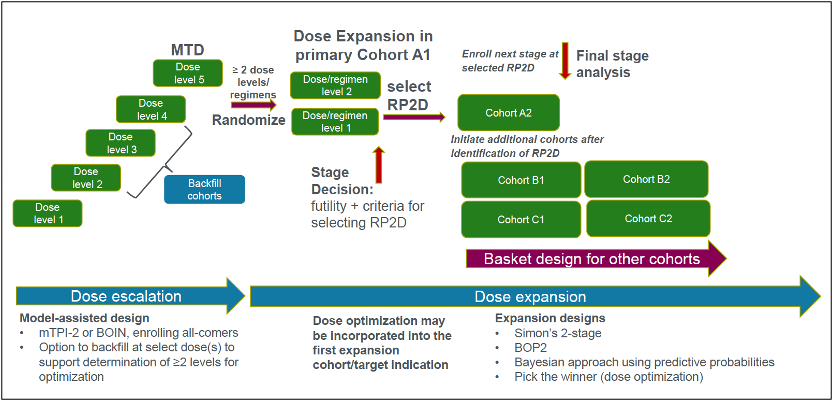
- Dose escalation: Model-assisted designs, such as the Bayesian Optimal Interval (BOIN) or Modified Toxicity Probability Interval (mTPI-2) design (Figure 1), are often preferred for dose escalation over traditional 3+3 designs, which can yield inaccurate and variable MTD estimates.4 It is operationally inconvenient to limit each cohort to three patients at a time, and there is a risk of missing the target by random chance or rule-based decision-making. In a BOIN design, there is no assumption about the dose-toxicity curve, and a transparent pre-calculated decision table makes it easy to escalate or de-escalate doses based on dose-limiting toxicities (DLTs). There is flexibility in target toxicity (versus the fixed rate of less than 33 percent in a 3+3 design) and in the number of patients per cohort (not limited to 3 or 6 patients). "Backfilling" cohorts at promising, safe dose levels allows for the collection of more data on safety and preliminary signals for antitumor activities.5 However, backfilling strategies require careful planning and cross-functional coordination to obtain preliminary evidence on the target validation, disease selection, and dose levels for the randomization dose-finding part.
- Dose optimization and expansion: At Parexel, we recommend including backfills during dose escalation to identify an efficacious dose range from which two or more dose levels are selected for the dose optimization part in a homogenous disease setting. The agency now typically requires a randomized comparison of at least two dose levels in dose optimization studies to eliminate any potential bias in dose selection based on safety and efficacy outcomes. However, trials do not need to be powered to demonstrate statistical superiority. At Parexel, we recommend that sponsors consider a formal expansion design for each dose cohort, such as the Bayesian Optimal Phase 2 (BOP2) design or Simon’s two-stage design with stopping rules to potentially select the optimal dose at an interim analysis (Figure 1).
Figure 1. Seamless Phase I/II trial design incorporating dose optimization