患者主導の医薬品開発のためのパレクセルのソリューション
患者主導の医薬品開発には、患者支援者との30分以上の面談や、ウェブサイト上での意欲をかき立てられるような文章以上のものが必要です。 それは開発の初期段階から始まり、その後のほぼすべての意思決定に影響を与える体系的なアプローチです。 弊社の患者支援部門は、スポンサーが患者さんのインサイトを活用し、複雑化する償還環境に対応し、リスクを軽減し、患者さんに適切な利益をもたらす商業的に実現可能なプロダクトを開発できるよう支援します。
Published on: Jul 7, 2025
8 min
パレクセルによるFDAが承認した希少疾病用医薬品の分析から得られた教訓
治験依頼者、規制当局、医療技術評価 (HTA) 機関、患者支援団体 (PAG) は、新薬が生活の質、症状、副作用に与える影響について患者さんから直接意見を聞くことを求めています。患者報告アウトカム (PRO) はそれを達成するための重要なツールです。PROは医師や第三者による修正や解釈なしに、患者さんの心理面や機能面を測定します。多くの企業はこの点を理解しています。時間と労力をかけてピボタル有効性試験でPROを収集し、そのデータをFDAに提出しているにもかかわらず、多くの場合、その情報は製品ラベルに記載されません。その結果、米国の医療提供者や患者さんは、この情報を容易に入手できません。なぜでしょう?関連するPROツールを選択し、規制当局にとって十分な厳密さでデータを収集および分析することが困難だからです。
この問題を理解するため、パレクセルでは2017年から2022年までに承認された164の新規希少疾患用医薬品と生物製剤に関するFDAの臨床審査文書を分析しました¹。その結果、治験依頼者の63% (103製品) がピボタル有効性試験中にPROデータを収集したにもかかわらず、ラベルにPROデータが記載されているのは製品の13% (21製品) のみであることがわかりました。
FDAが承認した希少疾患用医薬品 2017~2022年 (n=164)
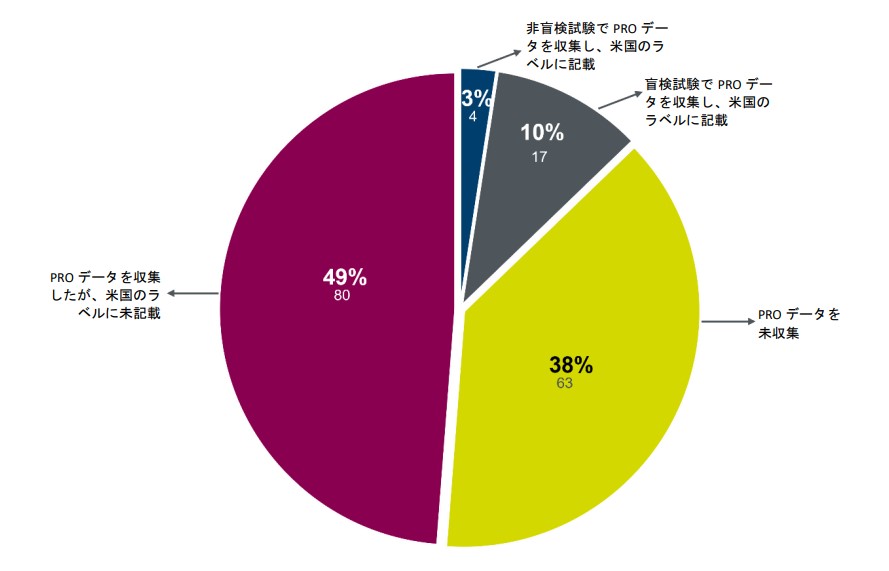
データセットの説明については、脚注1をご参照ください。
当然ながら、治験依頼者はさまざまな理由でPROを収集します。FDAはラベル交渉に関する情報を頻繁に削除するため、どのくらいの企業が正式な米国ラベル表記を求めているかを把握するのは困難です。FDAの審査担当者がラベルのPROデータを認めない場合でも、臨床審査文書ではそれを「裏付ける」エビデンスとして参照することがあります。
多くの場合、PROデータはFDA向けではありません。治験依頼者はEMAを目指している場合があります。最近の調査によると、EMAは「非盲検試験や健康関連の生活の質などの幅広い概念からのデータを受け入れる可能性が高い」ため、より多くのPROデータを製品ラベルに記載できるためです²。 治験依頼者によっては、主に欧州HTA機関の償還決定をサポートするためにデータを収集する場合もあります。
それにもかかわらず、希少疾患用医薬品に関するFDAの審査文書を分析したところ、少なくとも米国ではPROデータが効率的に利用されておらず、根強い方法論的な問題を抱えており、患者さんにもっと広く普及させる必要があることがわかりました。
患者さんが患者中心の医薬品開発の恩恵を受けるには、PROデータの開発・収集・分析のための技術と科学が進歩する必要があります。
弊社が分析した164の希少疾病用医薬品の承認に関する臨床・統計レビューや臨床結果評価 (COA) の協議において、FDAが頻繁に指摘したPROの問題には、次のようなものがありました。
PROは時には長い質問票に記入したり、紙や電子日誌に記録したり、すでに長時間に及んでいる訪問中に情報を提供する必要があるため、患者さんにとって負担となります。その結果、収集された後に解釈不能と判断されたPROデータは、患者さんの時間と労力の使い方が倫理的でないことを示しています。治験依頼者にとって理想的な戦略は、エビデンスに基づいた意義のあるPRO評価項目の構築に使用でき、患者さんの健康に関する高品質な測定データを特定することです。
パレクセルはPROの科学的厳密性を確保するために、3つのベストプラクティスを推奨しています。
PROツールには「内容の妥当性」が必須であり、研究対象の疾患に関連し、特異的でなければなりません。そのためには、疾患の自然経過、治療によって改善する可能性のある主な症状、臨床効果が得られるまでの適切な期間を明確に理解する必要があります。対象となる患者集団にとって重要であり、臨床試験において意義のある解釈可能な変化を示す可能性が高い、徴候、症状、影響などの概念を規定および定義します。患者さんや専門の医師からの意見を求めます。
これを適切に行うには、時間 (時には何年も) と労力が必要です。パレクセルでは現在、治験依頼者と協力して、小児のまれな中枢神経系障がいに対するPROをデザインしています。この疾患の臨床試験では、従来、発作の軽減を主要評価項目として採用しています。しかし、このような状況では、発作のコントロールだけが生活の質にとって重要な症状ではありません。これらの子どもたちは、ご家族や介護者、患者さんが重視する自分で食事を摂ることや、言葉を話すことができない場合が多いのです。そこで弊社では、自力での摂食と言語化の変化を定量化するための新しいPROを開発しています。患者さんや介護者さんと緊密に協力し、医学文献を徹底的に調査し、発声における意味のある変化とは何かと、それを正確に測定する方法を決定しました。すでに検証済みの複数のPROのモジュールを組み合わせて、この疾患で検証し、臨床試験で使用できる新しい評価ツールを開発しています。
最初は新しいPROを第I~II相試験の副次的評価項目、または探索評価項目として使用します。成功したら、これをピボタル試験に使用します。タイプBとタイプCのミーティング中に、このアプローチに関するFDAのガイダンスと承認を得られることを期待しています。新しいPROを開発するには、慎重な計画、強力な根拠、高品質のエビデンスが必要です。
治験依頼者は患者集団が有効かつ確実に自己報告できるようにし (患者さんによっては年齢が低すぎたり疾患が重度であるため行えない可能性があります)、さまざまな認知レベルと可動レベルに対応するPROを選択する必要があります。また、PROの想起期間は最大2週間になるようにしてください。これ以上長いとFDAによって、結果の妥当性を疑問視される可能性があります。
統計的妥当性と同等性を確保するために、PROデータの報告に使用されるメカニズムが試験全体を通じて一貫している必要があります。最近FDAによって承認された希少代謝性疾患の治療薬では、毎日の質問票を使用して患者さんの空腹レベルを測定しました。しかし、このピボタル試験の途中で患者さんは、紙から電子日誌による回答に切り替えられましたが、治験依頼者は新しい電子日誌デバイスのユーザビリティテストを行いませんでした。また、デバイスの機能、質問票の理解度、使いやすさを評価するための患者さんとの認知面接も実施しませんでした。FDAは質問票の中の1つの質問を指摘し、患者さんが理解していることを証明するにはデータが不十分であると結論付けました。この事例ではPROデータはラベルに記載されましたが (PROはこの試験の主要評価項目でした)、他の治験依頼者にとっては教訓となる話です。
2017年から2022年までの希少疾患用医薬品申請に含まれていたPROデータを解析して拒否したFDAの審査担当者は、規定の統計解析計画 (SAP) がないことや、第一種過誤 (偶然による結果) の修正を頻繁に指摘していました。この問題を回避する方法の1つは、ピボタル試験を開始する前にSAPについてFDAと意見を一致させることです。PROが副次的評価項目として指定されている場合、複数の比較用に調整されたアルファレベル (結果が偶然によるものではないことを示すために必要な有意性のレベル) を備えた階層的テストフレームワークで、結果の有効性を保証できます。
FDAが挙げたもう1つの重大な問題は、死亡、疾患進行、治験の高い離脱率などによるデータの欠落でした。症状の重い患者さんや治療効果が見られない患者さんは、治験が進むにつれて生活の質 (QoL)に関する患者報告ツールに記入しなくなり、結果が歪められます。重度の治療歴を持つ進行がんの患者さんを登録したある希少がん試験では、36項目の健康関連の生活の質 (HRQoL) 質問票の患者完了率がベースライン (約90%) から 48週目に急激に低下 (約30%) したため、データはFDAの審査担当者により無効と判断されました。
2022年のFDAの患者中心の医薬品開発 (PFDD) に関する会議で、FDAの上級臨床アナリストは、治験依頼者はデータが欠落している理由とその対処方法を理解するための計画を立てる必要があると指摘しました³。 治験依頼者は欠落データによる潜在的な偏りと解釈可能性の問題に対処するために、HRQoLデータが欠落している患者さんを異なる方法で管理する最悪のシナリオを想定した解析を計画することができます。パターン混合モデルなどのより洗練された統計手法を使うことで、欠落データが試験結果に与える影響をさらに明らかにできる可能性があります。
治験ではデータの欠落は避けられませんが、治験依頼者はそれを防ぐための措置を講じることができます。1回の治験で複数のPROの質問票や電子日誌に記入する負担が大きすぎると、患者さんは記入しません。そのため治験依頼者は試験デザインが理にかなっており、実践的であり、プロトコルで定められた目標を達成できることを確認する必要があります。
2017年から2022年までのPROデータを含む21の希少疾患用医薬品ラベルのうち、非盲検化のデータを報告したのはわずか4つ (19%) でした。
FDAが製品ラベルへの記載を承認したPROデータのほとんどは盲検化され、対照群を伴う研究で生成されました。
非盲検化や単群試験から得られたPROデータの解釈可能性は限られています。治療の割り当てを知っている患者さんによって、治療効果が意図的に過大評価または過小評価される可能性があるためです。
ランダム化比較試験 (RCT) は交絡因子を中和する最も効率的な方法であるため、リスクとベネフィットを調査する最良の方法です。治療と対照への二重盲検割り当てにより、既知の因子と未知の因子を公平に比較できます。ただし、顕著な活性が認められる細胞・遺伝子治療などの一部の治療法や、進行性がんや既存の治療法のない超希少疾患などの適応症では、比較対照群が実行不可能で倫理的でない場合があります。その場合は比較対照として、実薬対照、外部対照群、自然歴研究、既存対照を使用できます。
しかし、単群非盲検試験でも有益なPROデータを収集できます。希少疾患用医薬品の分析で、がんに対するCAR-T細胞療法をFDAが単群非盲検第II相試験に基づいて承認したことがわかりました。このピボタル試験では、副次的評価項目として3種類のPROを収集しました。PROデータは製品ラベルへの記載に関するFDAの基準を満たしていませんでしたが、データはEMAラベルに記載され、後に査読付き雑誌に掲載されました⁴。 雑誌に付随する論説には、「…このCAR-T細胞療法の負担に関する前例のないPRO情報を科学界に提供している点で、著者らの努力は称賛されるべきである。さらなる研究で解明されるべき未解決の疑問が複数あるが、この論文で報告された結果は非常に励みになるものであり、この分野における他の質の高い研究の取り組みを刺激することが期待される」と書かれています⁵。
このレビューから得た最も重要な教訓は、治験依頼者が患者さんや疾病の専門家と協力してPROを選択または開発し、ピボタル試験の前に規制当局と意見を一致させる必要があるということです。患者さんが経験した薬剤の効果をより広範に収集して公開することで、より総合的で患者さん中心の治療を進めることができます。
Contributing Expert
¹ Numbers include original new drug applications (NDAs) and biological license applications (BLAs) only, not supplemental applications (label extensions or new formulations). The dataset includes only orphan-designated products that achieved U.S. licensure for the first time between 2017 and 2022 and contained a new molecular entity or new active moiety. For CBER approvals, we excluded assays, fractionated plasma products, patch tests, reagents, vaccines, and tissue transplant products.