英国市場への参入を加速:MHRAの新国際承認手続きを活用
9 min
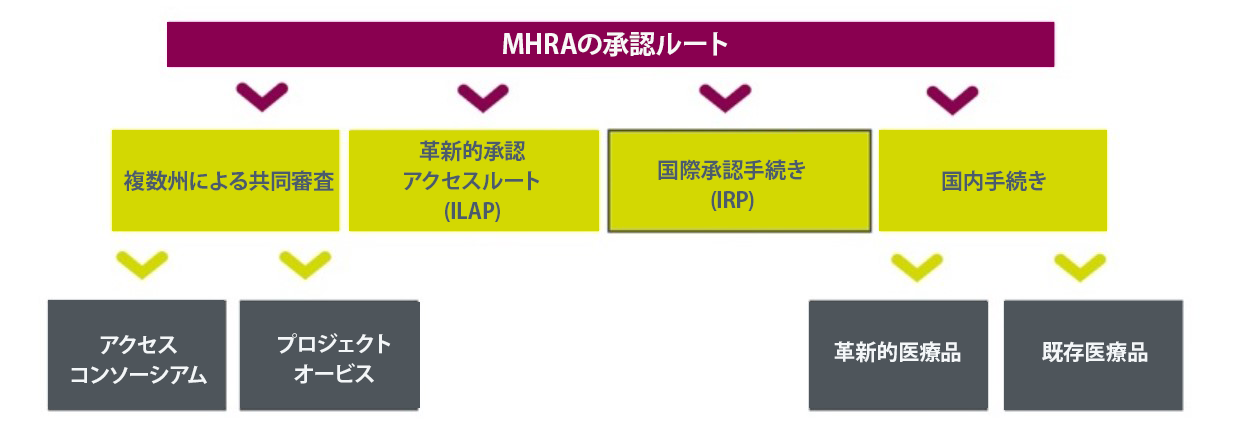
2020年初頭のEU離脱に伴い、英国は集中審査、分散審査、相互承認手続きといった欧州の規制ルートへの参加資格を失いました。これを受け、医薬品医療機器規制庁(MHRA)は、約7000万人の英国市場に製品を投入しようとする製薬企業向けに規制を緩和し、プロセスの効率化を図っています。MHRAの国際承認手続き (IRP)は、最近導入された複数の規制ルートの1つです。
*革新的な医療品については、150日での迅速承認の可能性があります。
図1. HMRAの承認ルート
IRPは英国の医薬品承認における重要な進展を示しています。このルートでは、MHRAが信頼できるパートナー機関の承認を認めることで、対象を絞った評価を実施し、特定製品の承認プロセスを迅速化できる可能性があります。これにより、英国市場への早期参入が期待できるだけでなく、企業はグローバル規制当局への申請順序を戦略的に見直し、より効率的で費用対効果の高い医薬品開発を行う機会も得られます。
IRP認定ルート:比較
対象要件:IRPはMHRAが指定する参照規制当局 (オーストラリア、カナダ、
EU (EMA) および加盟国規制当局、ノルウェー、アイスランド、リヒテンシュタイン、日本、スイス、シンガポール、米国) から既存の承認を取得している申請者が対象です。
所要期間:リスクと複雑性に関するMHRAの評価に基づき、二重ルート制度 (IRP承認AまたはB)が設けられています。承認A (60日間)と承認B (110日間)はいずれも、新規販売承認及びライン拡張にのみ適用されます 1。主な特徴は図2及び表1に示されています。
IRPのタイムラインは通常、英国国内手続きより短いですが、他規制当局の承認に依存します。
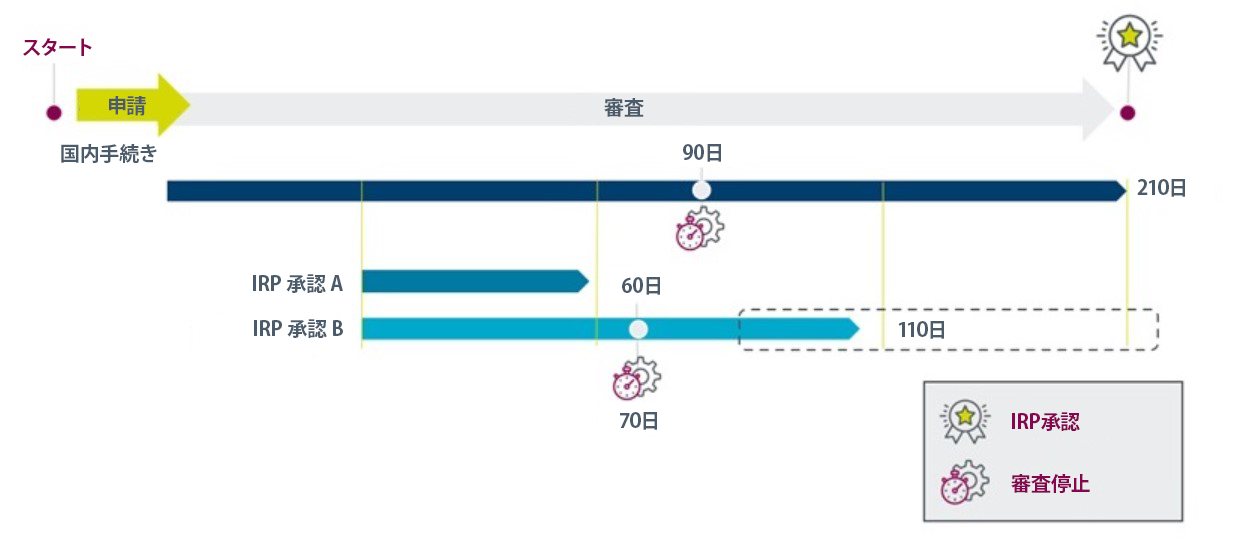 図2. 英国国内手続きとIRPのタイムライン比較
図2. 英国国内手続きとIRPのタイムライン比較
| 手続きの種類 | 承認A | 承認B |
| 商品の種類 | 標準医療品 | ATMP、ファースト イン クラスの新規有効成分、新規技術、分画血漿製剤に適用 |
| 参照規制当局での承認期間 | 過去2年以内に参照規制当局により承認された製品 | 過去10年以内に参照規制当局により承認された製品 |
| その他の基準 | 認定B基準を満たす製品は除外 | 希少疾病用医薬品指定、条件付き販売承認、例外的な状況、承認後安全性調査(PASS)を含む |
| CHM協議要件 | 医薬品委員会 (CHM)協議は不要 | CHMとの協議が必要 |
| タイムライン | 60日間スケジュール | 110日間スケジュール |
| 審査停止 | 審査停止なし | 70日目で審査停止 |
※網羅的リストではありません
表1. IRPにおける認定Aおよび認定Bの主要要素
IRP申請要件と範囲
- eCTD:EUフォーマットに準拠し、英国固有のモジュール1を含む必要があります。場合によっては一部改訂や編集が必要です。MHRAはさらなる改善を望んでおり、将来的には米国FDAの申請書類を受け入れる可能性も示唆しています。このテーマに焦点を当てた「今年実施予定のパイロット計画」も導入予定です 2,3。
- 名称に関する考慮事項:MHRA IRPガイダンスでは、英国販売承認において標準的なMHRA要件が適用されます。申請前にはMHRAの発明名称専用メールボックスに連絡し、フィードバックを得る必要があります。これは他地域で承認された名称と類似する英国国内ライセンスが存在する場合、MHRAが当該名称を承認しない可能性があるためです。安全性上の懸念に対応するため、代替の名称を提案する必要が生じる場合があります。
- 承認後手続きへの適用:初回申請に加え、IRPはライン拡張や変更(タイプ1B、タイプII)、更新申請(条件付きMAの年次更新、例外状況MAの年次再評価も含む)にも利用可能です。IRPは単独国内ルート、MRDCRP(多国間規制開発承認プロセス)ルート、もしくはECDRP(欧州共同開発承認プロセス)ルートを通じて、初回承認またはその後変更された製品のライフサイクル期間中に利用できます。IRPを通じて承認された製品は、単独の国内承認後の手続きを提出することも認められています 1。
企業がIRPから得られるベネフィット
MHRAの新たな承認ルートは、従来の米国NDA/BLAを申請した後、EMA、英国、スイス、日本の順で進めるというグローバル規制戦略の再構築の可能性を秘めています 4。FDA承認を活用することで、EUより先に英国で製品登録が可能になる場合もあります。日本、シンガポール、オーストラリアで既存製品認可を持つアジア太平洋地域の企業にとって、IRPは効率的な英国市場参入ルートとなるでしょう。
ブレグジットに伴う英国のEMA集中審査制度からの離脱により、IRPはその影響の一部を緩和する可能性もあります。英国承認はEU承認と比較的近い時期に得られるか、別の参照規制当局を活用すれば、より早期取得も見込めます。
MHRAは申請2~5年前からパイプライン会議を提供し、申請者の規制戦略策定を支援しています 2。
IRPルートのその他のメリットとデメリットは以下の通りです。
| メリット | デメリット |
| より的を絞った審査と短い審査期間。過去2年以内に参照規制当局によって承認された製品は特に有効 | 既存医薬品に対するジェネリック及びハイブリッド申請には、英国参照製品の入手可能性が必要 |
| 国内申請と比較した際のコスト削減が可能 | 全ての法的根拠が適用されるわけではありません (例:既存の使用方法は対象外) |
| EUと英国申請で同一書類 (英国固有のM1書類を除く)を流用可能なため、リソース負担が軽減 | 変更申請の審査期間は英国国内申請と同等 |
| 製品ライフサイクル全体にも適用可能であり、これにはライン拡張、変更 (タイプ1B、タイプII)、更新申請、ならびに単独の国内承認ルート、MRDCRP (多国間規制開発協力プログラム)、またはECDRP (欧州共同開発規制プログラム)を通じた当初の承認、もしくはその後変更された製品も含まれます。IRP (統合承認プロセス)を通じて当初承認された製品のみに限定されません。 |
表2. IRPの長所と短所
結論
IRPはMHRAの規制ツールキットを強化し、英国の患者さんへの医薬品アクセスを加速させると同時に、申請者に効率的な承認経路を提供します。過去の承認を活用することで、包括的な規制審査の必要性も軽減されます。MHRAの月次パフォーマンス報告書によると、主要な評価目標は着実に達成されています。
パレクセルではIRPを英国製品登録の主要な選択肢と位置付けています。
複数の規制分野に精通した元MHRA審査官を擁するパレクセルは、IRPをはじめとするMHRAの柔軟な制度を最大限に活用するために以下の支援を提供しています。
- 戦略的計画立案:可能な全ての選択肢を考慮し、製品に最適な規制経路を決定。英国申請がグローバルな規制戦略全体にどう適合するかも検討。
- 申請支援:IRP申請書の作成・提出、MHRAの全要件への準拠を確保。
- 規制情報:変化するMHRAの方針・手続きに関する最新情報を提供し、事前対応を支援。
- 包括的アプローチ:規制面だけでなく、英国およびそれ以上の市場アクセスや商業戦略への影響も考慮。
お気軽にお問い合わせください。いつでもご相談に応じます。
参考文献
- MHRA Guidance on International Recognition Procedure (updated 13 May 2025)
- Tasneem Fatima Keshavji (MHRA), RAPS Euro Convergence, 14th May 2025
- Pink Sheet, UK’s International Recognition Procedure: EU Dossier Faster Than US Or Canada, Eliza Slawther 15th May 2025
- Global Pharmaceutical and Biologics Regulatory Strategy, second edition. Chapter 7 CMC Regulatory Strategy. Antonelli S, Craig M. Regulatory Affairs Professional Society (RAPS) Publishing
ご注意:本情報は教育目的のみで提供するものであり、法的または規制上の助言として解釈されるべきものではありません。本情報をもとに行動される際は、必ず専門家の助言をお受けください。